





Familial hypercholesterolaemia (FH) is an autosomal-dominant disorder associated with mutations in the LDL receptor (LDLR) gene resulting in markedly elevated plasma low-density lipoprotein cholesterol (LDL-C) levels and premature atherosclerotic cardiovascular disease (ASCVD). Heterozygous FH (HeFH) (mutation in one allele) is associated with plasma LDL-C levels >190 mg/dl, whereas homozygous FH (HoFH) (mutation in both alleles) is associated with plasma LDL-C levels >500 mg/dl.1,2 As a result, there is a 20-fold increase in risk of premature coronary heart disease (CHD) in untreated patients compared to control.3 HeFH patients usually develop CHD before age 55 and 60 respectively for men and women without treatment.4 HoFH patients, however, develop CHD very early in life and can die before age 20 without treatment.4
In general, an estimated 20 million people worldwide have FH.4 Approximately 1 in 300–500 people have HeFH and 1 in 1 million have HoFH.3 In certain populations, such as French Canadians and Dutch Afrikaners, the prevalence is as high as 1 in 100.3 Despite the high prevalence and increased risk of premature ASCVD in untreated patients, less than 1 % are diagnosed with FH worldwide.4
Family history, physical examination and a lipid profile are essential to establishing a diagnosis of FH. FH should be suspected with fasting LDL-C ≥190 mg/dl in adults and ≥160 mg/dl in children if secondary causes of hypercholesterolaemia such as hypothyroidism, nephrotic syndrome and liver disease are ruled out.2,5 The clinical diagnosis of FH can be established by five criteria: 1) the presence of early coronary artery disease (CAD) in the index case; 2) family history of premature CAD; 3) elevated LDL-C; 4) tendon xanthomas; 5) corneal arcus.2 In addition, validated clinical tools such as US Make Early Diagnosis Prevent Early Death (MEDPED), Dutch Lipid Clinic Network and Simon- Broome Registry can assist in the diagnosis of FH.5
Although genetic screening is not necessary for the diagnosis and clinical management of FH (studies report 5–20 % of FH patients are without identifiable mutation), it can augment screening strategies and help predict cardiovascular risk if a specific genetic anomaly can be identified.5,6,7 In a large Dutch cohort of nearly 30,000 individuals, finding a pathogenic LDL receptor mutation nearly tripled the cardiovascular risk for that patient (HR 3.64, CI 3.24–4.08, P<0.001).6 Data also show improved sensitivity of a genetic diagnosis compared to one based on epidemiologic LDL distributions. In a study of over 26,000 individuals in the Netherlands, an LDL above the 90th percentile provided a 68.5 % sensitivity while an LDL receptor mutation genetic screen demonstrated a sensitivity of 91.3 %.8 Once an accurate diagnosis is made of FH, cascade screening of all firstdegree relatives is paramount.
Treatment Guidelines
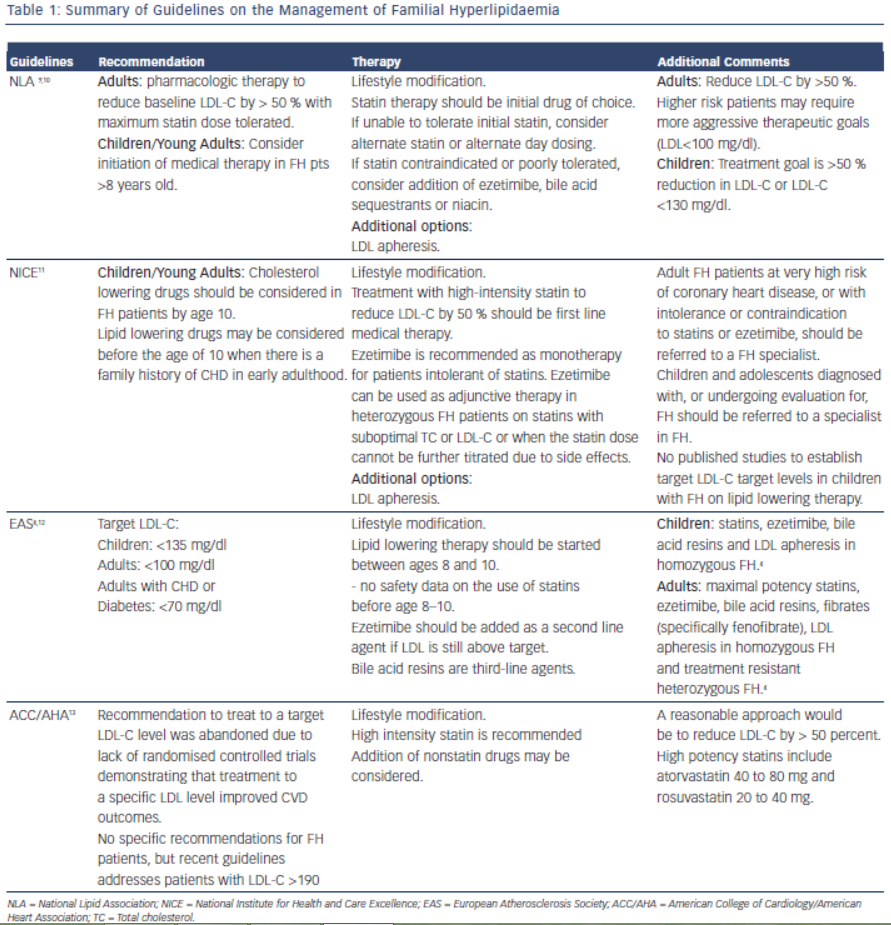
Early detection and treatment of FH is recommended to reduce risk of cardiovascular events. The goal of treatment is to reduce LDL-C by 50 % from baseline levels.9 This was based on two trials which showed reduction in CIMT (Carotid Intimal Medial Thickness) in patients who achieved > 50 % reduction of LDL-C on statin therapy.10,11 Lifestyle modification, lipid-lowering therapy, LDL apheresis and in rare cases, liver transplant are the cornerstones of LDL-C lowering in patients with FH. Table 1 summarises the current guidelines for the treatment of FH.
It is important for the clinician to be aware of the various treatment options for FH, as combination therapy will likely be required to achieve goal reductions in LDL-C. Investigators have reported as many as 77 % of patients with an FH diagnosis and on cholesterol medication do not achieve goal LDL-C levels within two years of diagnosis.12 The most common reason cited (32 %) for inadequate control of the patients’ cholesterol levels was “acceptance” of a suboptimal LDL-C level by a treating physician.13 Other causes of inadequate control include improper dosing of medications and late age of diagnosis.14
Lifestyle Modifications
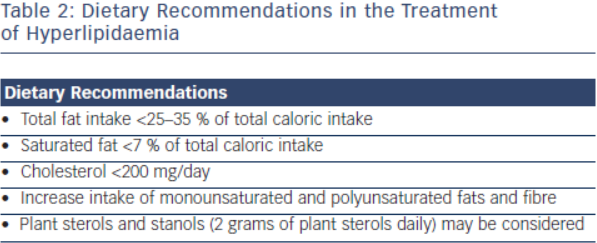
Lifestyle modification is the foundation in the treatment of FH. Primordial prevention which includes counselling for smoking, avoidance of diabetes and regular physical activity is an integral component for treatment in this population.1 Dietary recommendations from the National Lipid Association and International FH Foundation include are outlined in Table 2.15 These groups’ recommendations also emphasise limiting alcohol intake (one drink daily for women, 1-2 drinks daily for men) and tobacco cessation.16 Referral to appropriately qualified nutritionists and dieticians is also recommended.2 Daily physical activity is encouraged, and achieving an optimal weight for overweight patients should be stressed. For every 10 kg of weight lost, there is an 8 mg/dl reduction of LDL-C.17,18 While lifestyle modification is the first step in the treatment of FH patients, this strategy alone will be insufficient as the sole treatment to achieve LDL-C goal. Lifestyle modification will at most reduce LDL-C concentration by 10–15 %19 and most patients will require further pharmacological therapy.
Pharmacologic Treatment
Patients with LDL >190 mg/dl are identified within the new ACC/ AHA 2013 Blood Cholesterol Guidelines as requiring pharmacologic intervention.20 The required treatment regimen and the individual’s response to this treatment varies according to the genetic and phenotypic heterogeneity of FH, with autosomal recessive hypercholesterolaemia responding more robustly to treatment.1
Statins
Statins inhibit hydroxyl-methylglutaryl coenzyme A reductase (HMGCoAR), a key enzyme in the synthesis of cholesterol in the liver, ultimately decreasing LDL production. Statins also lead to up-regulation of LDL receptors; however this effect is limited due to enhanced compensatory degradation of such receptors. In patients with HoFH, the effectiveness of statins is attenuated given its mechanism of action involves up-regulation of liver LDL receptors. This is the case also for ezetimibe and bile acid sequestrants.21
Statins are first line pharmacologic therapy agents in the treatment of FH; they reduce cardiovascular mortality even in receptor-negative patients.22 Current guidelines recommend maximal statin therapy to reduce LDL-C levels by at least 50 % from baseline.20,23–25 Therapy should be started with a high potency statin at the greatest tolerated dose. Plasma levels of hepatic transaminases, creatine kinase (CK), glucose and creatinine should be measured before starting drug therapy.
Despite aggressive statin therapy, in the majority of patients with FH, even the highest doses of the most potent statins do not yield more than a 50 % LDL-C reduction. In these patients a combination of lipidmodifying agents enhances efficacy.3,21,22,26–28 Most often, the cholesterol absorption inhibitor Ezetimibe is chosen as second line therapy.3
Cholesterol Absorption Inhibitors
Ezetimibe is a selective cholesterol absorption inhibitor that blocks the absorption of dietary cholesterol and delivery of intestinal cholesterol to the liver resulting in up-regulation of hepatic LDL receptors and enhanced clearance of LDL particles from the circulation. Statins and ezetimibe taken together potentiate the LDL reduction. This reduction in LDL-C has been documented between 10–40 percent. The variable response is likely due to differences in the amount of residual functional LDL receptor activity, with those patients with no receptor activity (receptor negative) having limited response to drugs that work through mechanisms involving LDL receptor up-regulation. In one report, ezetimibe added an extra 15 % reduction in LDL levels when added to rosuvastatin 40 mg.29 Despite the added effect of ezetimibe, many patients require further LDL lowering therapy, including bile acid sequestrants, to achieve therapeutic goals.
Bile Acid Sequestrants
Bile acid sequestrants bind cholesterol in the gut thereby enhancing cholesterol excretion and reducing the enterohepatic circulation of bile acids and biliary cholesterol. The reduction in bile acids leads to a compensatory increase in conversion of cholesterol to bile acids, up-regulating expression of hepatic LDL receptors and consequently increasing hepatic removal of LDL particles from the circulation.30 Bile acid sequestrants, specifically colesevelam (the newest in its class), have been shown to reduce LDL-C levels alone and more significantly when used in combination with ezetimibe and statins. The combination of these drugs was proven to be safe and well tolerated.31–33 In FH, bile acid sequestrants are considered second line therapy after treatment with statins and ezetimibe.34
Niacin
In patients with FH, niacin is considered an alternative agent for patients after treatment with statins, ezetimibe and bile acid sequestrants. It can be used to further intensify therapy or as replacement when statins are not tolerated.3,35 Its beneficial effects on the lipid profile include increasing HDL-C by 15-30 %, lowering triglycerides up to 35 % and lowering LDL-C about 20 percent.36 Despite these favourable effects, two recent studies showed lack of clinical significance in terms of cardiovascular outcomes. It should be noted however that these two studies were not designed specifically to address the benefit of niacin as adjunct therapy for patients with FH. The recent Atherosclerosis Intervention in Metabolic Syndrome with Low HDL Cholesterol/High Triglyceride and Impact of Global Health Outcomes (AIM-HIGH) study showed that despite a significant increase in HDL, niacin did not reduce cardiovascular events compared to placebo when added to simvastatin.37 Similar results were demonstrated in the Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) trial in which more than 20,000 patients with known vascular disease were randomised to placebo or niacin in addition to statin or statin/ezetimibe therapy. The trial was stopped prematurely since niacin did not further reduce the risk of the composite endpoint and was found to increase the incidence of nonfatal serious side effects.38
Newly FDA Approved Agents, Mipomersen and Lomitapide
In the last year, two new drugs have been approved by the US Food and Drug Administration (FDA) as adjunct therapy for FH: mipomersen (an apolipoprotein-B [apoB] synthesis inhibitor) (Kynamro–Genzyme) and lomitapide (a microsomal transfer protein inhibitor) (Juxtapid– Aegerion).39 These agents interfere with the production and secretion of apoB-containing lipoproteins respectively, independent of the LDL receptor. These agents are used after statins, cholesterol absorption inhibitors and bile acid sequestrants have been considered.
Mipomersen
Mipomersen is an antisense molecule targeting the messenger ribonucleic acid (mRNA) that encodes apoB-100 produced by the liver. ApoB-100 is the main structural protein of LDL, and its precursor, VLDL. Mipomersen has been tested in four populations: 1) HoFH, 2) HeFH with coronary artery disease, 3) Patients with severe hypercholesterolaemia and 4) Patients at high risk for CAD with hypercholesterolaemia.40–43 All trials were phase 3, randomised, double blind, placebo-controlled and multicentre studies. Mipomersen demonstrated statistically significant reductions in LDL and apoB as a single drug or in combination with statins and other agents.40–45 It is administered weekly by subcutaneous injection with rapid absorption and distribution to tissues. The most common adverse effects observed in clinical studies were erythema or pain at the injection site, which were self-limited, flu-like symptoms and transient elevations in CRP at the beginning of the injections, with no other inflammatory marker elevations. Elevations in hepatic transaminases were less common, largely reversible with no other liver function tests abnormalities or clinical implications. Increase in liver fat was also observed, however it stabilised after a year of treatment. In combination with statin, mipomersen showed a LDL reduction of approximately 25 percent.40,46
Lomitapide
Lomitapide binds and inhibits the microsomal triglyceride transfer protein (MTTP). MTTP is critical in the synthesis of cholesterol particles, transferring triglycerides onto apo B in the hepatocyte to form VLDL and in the enterocyte to form chylomicrons.47 Lomitapide is given orally in doses of 5–50 mg daily and is rapidly absorbed with a significant first pass effect in the liver. It is metabolised by CYP3A4 and therefore has the potential for drug interactions with commonly prescribed medications. Ketoconazole can significantly increase levels of lomitapide while simvastatin and warfarin are both increased in concentration when coadministered with lomitapide.48 Gastrointestinal intolerance is the most common adverse effect with patients reporting diarrhoea, nausea and flatulence. These side effects can be ameliorated by slow dose uptitration and adherence to a low fat diet. Vitamin E deficiency is a potential adverse effect. Given patients with FH have higher baseline levels of vitamin E, treatment with lomitapide often results in a normal vitamin level.46
Similar to mipomersen, lomitapide can asymptomatically increase serum transaminases in some patients with no changes in bilirubin and alkaline phosphatase. Transaminase elevations appear to be reversible and transient. Mild increases in liver fat content have been observed and could potentially be counteracted by following a low fat diet.49 The long-term consequences of this induced hepatosteatosis remain unclear and will require future investigation.
Mipomersen and lomitapide were approved with an enclosed warning describing the risk of hepatotoxicity. Due to the overall risk profile, these medications are available only through the restricted Kynamro/ Juxtapid Risk Evaluation and Mitigation Strategy (REMS) Program. Only certified health care providers can prescribe the medicines and only certified pharmacies may dispense them. Risks considered, mipomersen and lomitapide should be considered as adjunctive therapy to diet and cholesterol-lowering drugs in adults with homozygous FH to further reduce plasma LDL cholesterol, particularly if lipoprotein apheresis (LA) is not available.50
PCSK9 Inhibitors
Several new medical therapies are being evaluated in clinical trials for the treatment of FH. The most promising of which are the PCSK9 inhibitors, a completely novel class of medications. PCSK9 is a serine protease that is involved with the degradation of LDL receptors leading to higher circulating plasma LDL-C levels. PCSK9 inhibitors are injectable monoclonal antibodies that bind to PCSK9, thereby decreasing the turnover of LDL receptors, which in turn leads to a decrease in circulating LDL-C levels. Two of the most promising medications in this novel class are evolocumab and alirocumab. Both of these medications have undergone extensive phase II trials and are in the process of completing phases III trials. The DESCARTES phase III trial recently presented 52-week follow up data that compared evolocumab and placebo in patients already treated with standard lipid lowering therapies.51 The investigators’ data show a 57 % reduction in LDL-C in the evolocumab group compared to placebo.
Further supporting this class of medications is the ODYSSEY trial, which randomised heterozygous FH patients on standard lipidlowering therapy to receive alirocumab or placebo.52 The results of this trial showed that alirocumab decreased LDL-C by 49 % from baseline while the placebo group had a 9 % increase in LDL-C. Impressively, in this traditionally very difficult to treat FH population, the percentage of patients reaching goal LDL-C was 70–80 %. Moreover, the post hoc analysis of a separate ODYSSEY Long Term trial was notable for a 54 % reduction in major adverse cardiovascular events in the alirocumab group versus placebo.53 This result was statistically significant.
There are several other medications being evaluated for the treatment of FH but none seem to be as promising as the PCSK9 inhibitors. The MODE trial is a phase II examination of recombinant ApoA1 milano infusions in homozygous FH patients.54 Finally, anacetrapib, a CETP-inhibitor is being studied in homozygous FH patients in a phase III trial.54
Lipoprotein Apheresis
When lifestyle modifications prove inadequate and pharmaceutical interventions are either not tolerated or lack adequate response, lipoprotein apheresis (LA) can be used to remove circulating LDL along with other atherogenic particles. All apheresis processes require the removal of blood from a vein (typically the antecubital) and typically involve the separation of that blood into cellular and plasma components. LA sessions typically last three hours and are performed on average every two weeks for HeFH patients and 7–10 days. Hyperlipidaemia HoFH patients.55
Once the blood is removed, several strategies can be employed to remove atherogenic compounds from the plasma including immunoadsorption, plasmapheresis, direct adsorption, dextran sulfate adsorption, and heparin extracorporeal LDL apheresis. Plasmapheresis was one of the original methods used to remove LDL from serum. During this process, plasma is separated from the cellular components of the blood and is removed from the body and replaced by albumin infusions. This process was non-selective and removed helpful plasma protein including HDL and immunoglobulins. Immunoadsorption is more selective for the atherogenic particles. In immunoadsorption, the plasma is passed over membranes coated with polyclonal sheep antibodies against apolipoprotein B100 and apolipoprotein B containing lipoproteins thereby removing LDL, VLDL and lipoprotein(a).
Not requiring separation of the plasma and cells, direct lipoprotein adsorption employs polyacrylamide beads within a filter. These beads contain pores of a particular diameter that allows only certain particles to enter and thus be removed from the patient’s serum. Based on the width of the pore, the filters can be more or less selective which is typically determined by disease severity. Typically, these columns can remove LDL, lipoprotein(a) and triglycerides but can also remove fibrinogen in the more non-selective columns.
Dextran sulfate adsorption (DSA) involves separated plasma being passed over two cellulose-bead columns, which contain dextran sulfate. This interaction leads to electrostatic binding of (apolipoprotein- B)-containing particles and their removal from circulation. This process, similar to the above-mentioned immunoadsorption, is specific to apolipoprotein-B and does not remove HDL or albumin from circulation. DSA is the most efficient method to remove atherogenic particles from circulation given the quantity of plasma it can filter in a given session. Patients receiving DSA must be anticoagulated with heparin during the session, making heparin hypersensitivity a contraindication to DSA.
The last filtration process, heparin extracorporeal LDL apheresis (HELP), involves the heparinisation and acidification of the patient’s plasma, causing heparin to carry a negative charge and LDL to carry a positive charge. These opposite charges lead to heparin-LDL complex formation, precipitation and eventual filtration. Via this method 60–64 % of LDL, 65–75 % of VLDL and 60-70 % of lipoprotein(a) is removed.55–57
Given the relatively low numbers of patients receiving LA, few randomised clinical trials have been performed to examine its effectiveness.56 The Familial Hypercholesterolaemia Regression Study examined 39 patients randomised to LA and simvastatin or simvastatin and colestipol and found biweekly LA and simvastatin decreased LDL cholesterol levels by 31 % more than the medication only group.58 Similarly, the LDLApheresis Atherosclerosis Regression Study (LAARS) found reductions in the LDL levels of their 42 study subjects randomised to LA.59 Neither study showed a significant difference in coronary atherosclerosis as determined by coronary angiography.55,58,59 Other randomised studies have shown reductions in circulating inflammatory biomarkers such as CRP and lipoprotein-associated phospholipase A2.60,61
Given its association with improved LDL control, LA is approved in the United States for HoFH patients with LDL ≥ 500 mg/dL or HeFH patients with LDL ≥ 300 mg/dL or ≥ 200 mg/dL with documented coronary artery disease.55
Liver Transplantation
Orthotopic Liver transplantation (OLT) remains the only curative option for patients with homozygous FH that cannot achieve optimal LDL levels after pharmacological treatment and for those unable to tolerate lipid apheresis.19,50 Liver transplantation replaces dysfunctional hepatic LDL in receptors patients, allowing near normalisation of lipoprotein metabolism.62 While lipid apheresis can reduce or maintain LDL levels at acceptable ranges, it does not prevent development of atherosclerosis.63 The first liver and heart transplant64 for homozygous FH occurred in 1984 in a six-year-old female with severe coronary artery disease. The patient showed significantly improved and sustained lower LDL levels postoperatively, but ultimately died due to cardiac allograft rejection. Since then, over 30 cases with patients ranging from 2–46 years of age have been reported.64–67 The long-term outcomes of OLT are unknown due to paucity of donors, high-risk postsurgical outcomes, and the concern for life-long immunosuppressive therapy. However, a recent update has shown the five-year survival rates after OLT in paediatric patients approaches 90 % in the United States.68 Case reports have shown patients free of coronary artery disease 20 years after transplant, while others have had post-operative complications, immediate organ rejection, and complications from immunosuppressant therapy.62,63,65 Patients who are considered for OLT usually have severe coronary artery disease and valvular insufficiency, which requires simultaneous heart and liver transplant, further increasing postoperative risks and complications. Recent literature supports transplantation in a younger population before the onset of significant coronary artery disease to preclude the need for cardiac transplantation.66–67 Patients with homozygous FH who fail medical therapy and apheresis should be considered candidates for OLT.
Special Populations
While statins are the most effective therapy for those with FH, the use of statins is contraindicated during pregnancy. Animal studies have demonstrated conflicting evidence on the teratogenicity of statins during pregnancy.69 There have been studies that have found the lipophilic class of statins (lovastatin, cerivastatin, and fluvastatin) to be associated with skeletal malformations. Atorvastatin and lovastatin have also been associated with developmental toxicity and skeletal defects, but only at supratherapeutic doses that also caused maternal toxicity.69
There is limited data on the teratogenicity of other lipid-lowering agents in humans. Ezetimibe, nicotinic acid, and fibrates have all been associated with teratogenic effects in animal studies.69 Therefore, the only medications currently acceptable to use during pregnancy are the bile acid binding resins, cholestyramine and colsevelam, as these medications do not pass into the systemic circulation and have not been shown to have any adverse effects thus far.70,71
The National Institute of Clinical Excellence (NICE) guidelines recommend that all women stop taking statins three months prior to attempting to conceive.72 Women who become pregnant while taking a statin or other systemically absorbed lipid-modifying agent, should be instructed to stop treatment immediately and be referred to an obstetrician for urgent fetal assessment.73 Women should not resume statin therapy until after they have completed lactation.72
Conclusion
The treatment of FH has evolved considerably with the availability of novel potent LDL-C reducing agents such as lomitapide and mipomersen in addition to plasma lipoprotein apheresis. FH continues to be “hidden in plain sight” with a significant number of children and adults that remain undiagnosed. Initiatives such as the FH foundation and FH registry (http://thefhfoundation.org/) provide a great opportunity to raise awareness and increase the number of patients that are diagnosed and treated with FH.